转录组分析
转录组分析
为了寻找表型变化背后的分子机制,可能是一个基因,更可能是多个基因组成的相互作用网络共同导致了表型的差异 在DNA层次上也许是SNP、InDel、SV、CNV、甲基化等造成的差异。
在RNA层次上也许是差异表达、可变剪切、修饰等造成的差异。
在蛋白层次上也许是蛋白的丰度差异、折叠方式差异等造成的差异。
也可能是以上这些层次之间相互影响造成的变化
为什么研究RNA层次,一般蛋白上出现差异通常可以从RNA层次上可以看出来。
DNA的变异也可能在RNA的表达上表现出来
得到表达数据挖掘三张表
样本信息表
表达矩阵
基因信息表
在植物生物信息挖掘中,特别是在进行RNA测序(RNA-seq)后得到的表达数据,通常会涉及三张表:样本信息表、表达矩阵和基因信息表。这些表格提供了关于样本、表达量和基因的重要信息,对于进一步的分析和解释至关重要。 1.样本信息表: 样本信息表是描述每个样本的属性和特征的表格。它包含每个样本的唯一标识符(如样本ID或名称),以及与样本相关的各种信息,如处理条件、生理状态、时间点等。样本信息表提供了实验设计和样本分组的基础,帮助研究人员在后续分析中考虑到潜在的批次效应或其他实验条件对表达量的影响。 2.表达矩阵: 表达矩阵是一个二维矩阵,其中行表示基因,列表示样本,每个元素表示相应基因在对应样本中的表达量。表达矩阵是从RNA-seq数据中计算得出的主要结果之一。它反映了每个基因在不同样本中的表达水平,可以用于比较不同基因的表达模式、寻找差异表达基因以及进行聚类和分类等分析。表达矩阵的值通常是基因表达的定量值,例如FPKM(Fragments Per Kilobase of transcript per Million mapped reads)或TPM(Transcripts Per Million)。 3.基因信息表: 基因信息表提供了每个基因的注释和相关信息。它包含基因的唯一标识符、基因名、基因描述、功能注释、基因本体论述(Gene Ontology,GO)注释等。基因信息表提供了对于表达矩阵中的基因进行功能注释和解释的基础。通过基因信息表,研究人员可以了解每个基因的功能、参与的生物过程以及可能的调控机制,从而进一步理解表达数据的生物学意义。 这三张表在植物生物信息挖掘中是相互关联的。样本信息表提供了实验设计和样本分组的基础,表达矩阵提供了基因在样本中的表达水平信息,基因信息表提供了对基因功能和注释的解释。通过整合和分析这些表格的信息,研究人员可以揭示植物基因表达的模式、寻找差异表达基因、发现关键的生物学通路以及探索植物的生物学机制。
当研究人员想要了解哪些基因在特定条件下被表达、表达量如何变化时,他们可以使用两种常见的方法:表达芯片和RNA测序(RNA-seq)。
表达芯片是一种基于杂交技术的高通量基因表达分析方法。它使用已知的DNA探针序列来检测样本中特定基因的表达水平。首先,研究人员将待检测的RNA样本转录成互补DNA(cDNA),然后将其与芯片上的DNA探针进行杂交。芯片上的DNA探针代表了大量的基因,每个探针对应一个特定的基因。如果待测的RNA中含有与DNA探针互补的序列,它们会结合在一起。之后,通过检测结合的信号强度,可以确定基因的表达水平。表达芯片适用于检测已知基因的表达情况,但对于未知基因或基因组中的变异不太敏感。
相比之下,RNA测序(RNA-seq)是一种更先进、更全面的方法,可以提供更详细的基因表达信息。RNA-seq利用高通量测序技术,直接对RNA样本进行测序。首先,RNA样本被逆转录成cDNA,然后进行文库构建和测序。测序得到的是cDNA的序列信息,然后使用计算方法将这些序列比对到参考基因组上,以确定每个基因的表达水平。与表达芯片不同,RNA-seq不受限于已知基因,可以发现新的转录本、识别未知基因和变异,并提供更准确的表达量测量。此外,RNA-seq还可以提供转录本的边界信息、剪接变体和基因表达的动态范围。
因此,表达芯片主要适用于检测已知基因的表达水平,而RNA-seq可以提供更全面的信息,包括已知和未知基因的表达,并提供更详细的转录本分析。RNA-seq通常在研究中更受青睐,因为它具有更高的灵敏度和更广泛的应用范围,尤其适用于探索未知基因或进行深入的基因表达研究。
表达芯片和RNA-seq在敏感性和动态范围方面存在一些区别,这些区别主要源自它们的原理和技术差异。
敏感性: 表达芯片的敏感性通常较低。它使用预先设计的DNA探针来检测已知基因的表达水平,这些探针通常是特定的序列片段。因此,对于未知基因或未覆盖的基因,表达芯片无法提供相应的表达信息。此外,由于杂交过程和检测方法的限制,表达芯片可能无法准确地测量低表达基因或表达量变化较小的基因。
相比之下,RNA-seq具有更高的敏感性。它使用高通量测序技术直接测序RNA样本,因此不受预先设计的探针的限制。RNA-seq能够检测到低表达基因和罕见转录本,因为它能够检测到更广泛的RNA序列。此外,RNA-seq的信号强度与基因的表达量呈线性关系,使其能够准确测量表达量的变化。
动态范围: 动态范围是指可以检测到的最低和最高表达量之间的差异范围。表达芯片的动态范围通常较窄。由于使用的DNA探针是预先确定的,表达芯片对于高表达基因可能存在饱和效应,即高表达基因的信号强度达到上限,无法准确测量其真实的表达水平。另一方面,对于低表达基因,表达芯片的信号可能混杂在背景噪声中,导致较低的动态范围。
RNA-seq具有较宽的动态范围。通过测序RNA样本并计数每个基因的测序reads数量,RNA-seq可以提供基因表达的直接定量信息。由于没有信号饱和的限制,并且使用计数数据来表示表达量,RNA-seq能够捕获从极低到极高表达水平的基因,并提供更广泛的动态范围。
这些差异主要是由于表达芯片和RNA-seq的技术原理不同所致。表达芯片使用杂交技术和预先设计的DNA探针,而RNA-seq使用直接测序RNA样本的方法。RNA-seq的直接测序和计数优势使其在敏感性和动态范围方面具有更高的性能,尤其适用于研究未知基因或探索表达水平变化较大的基因。
- 比对
hisat2
把rna数据比对到参考基因组上
表达定量
标准化
TPM------------------------------------TMM
差异表达
log2FC FoldChange
生物学重复-----------------------多个生物学重复
测3个生物学重复
假设检验
统计分布----t检验
组内差异小 组间差异大-------------被判定为差异表达
如何筛选差异基因?
使用Pvalue adjust < 0.05认为是差异表达基因 Pv不能设置为0.02
生物学重复越多,鉴定出的差异基因就越多?
数据太少无法判断,但是数据多了可以增大决策的准确性
fold change >2 log2FC > 1
富集分析
样本聚类分析和WGCNA
相关系数:比较组织之间的重复
3*4*2
特征表达分析
共有和特有表达基因
01 研究背景
为了解释两种苹果果皮颜色的差异(一红一黄),已有研究表明和花青素的合成与积累有关。
目前,花青素的合成和积累受到三个方面的影响。在合成方面,苹果中的花青素合成酶大多与积累呈正相关,而葡萄中只有少数酶与积累相关。在调控方面,转录因子bHLH、WD40和MYB对花青素的调控至关重要,其中MYB因子发挥着关键作用。在运输和存储方面,花青素从细胞质转运到液泡中,液泡中的存储则由谷胱甘肽S转移酶(GSTS)和多药和有毒化合物外排转运蛋白(MATES)负责。
这两种苹果在花青素合成与积累有什么差异?能否找到差异的基因,差异基因的表达又是怎么样造成的(DNA变异?表观修饰?转录调控?),能否找到影响差异的分子机制?
这篇文章的样品与测序情况:
5年生 苹果树 3 个生物学重复,2个品种,4个时期(S1: 7月16日; S2: 8月7日; S3: 8月26日; S4: 9月14日),共 24 个样本。 模式: Single-end 读长: 101 bp 测序仪: Hiseq 2000
02 分析案例
(1)寻找测序数据

SRA高通量数据
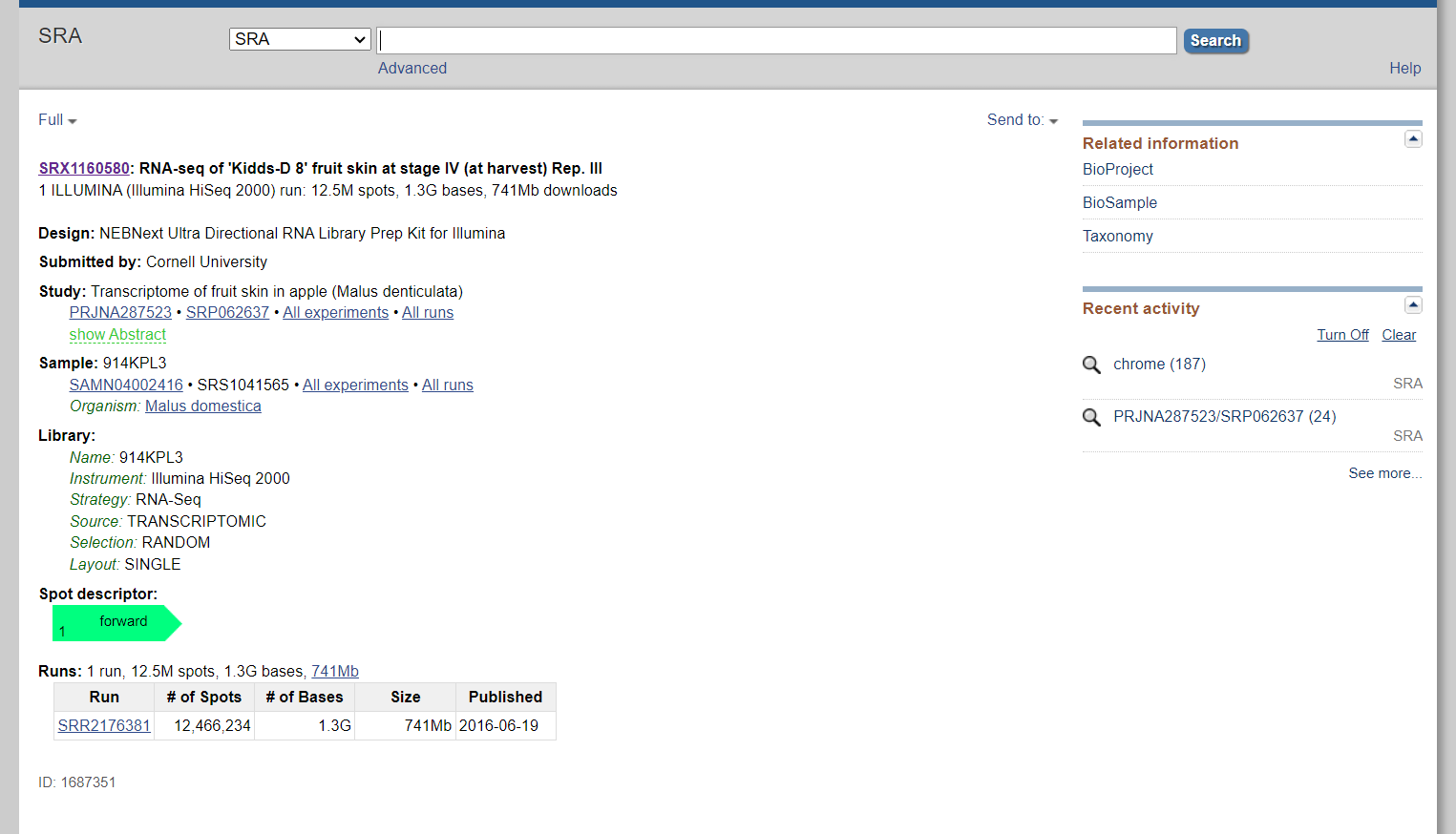
1 | |
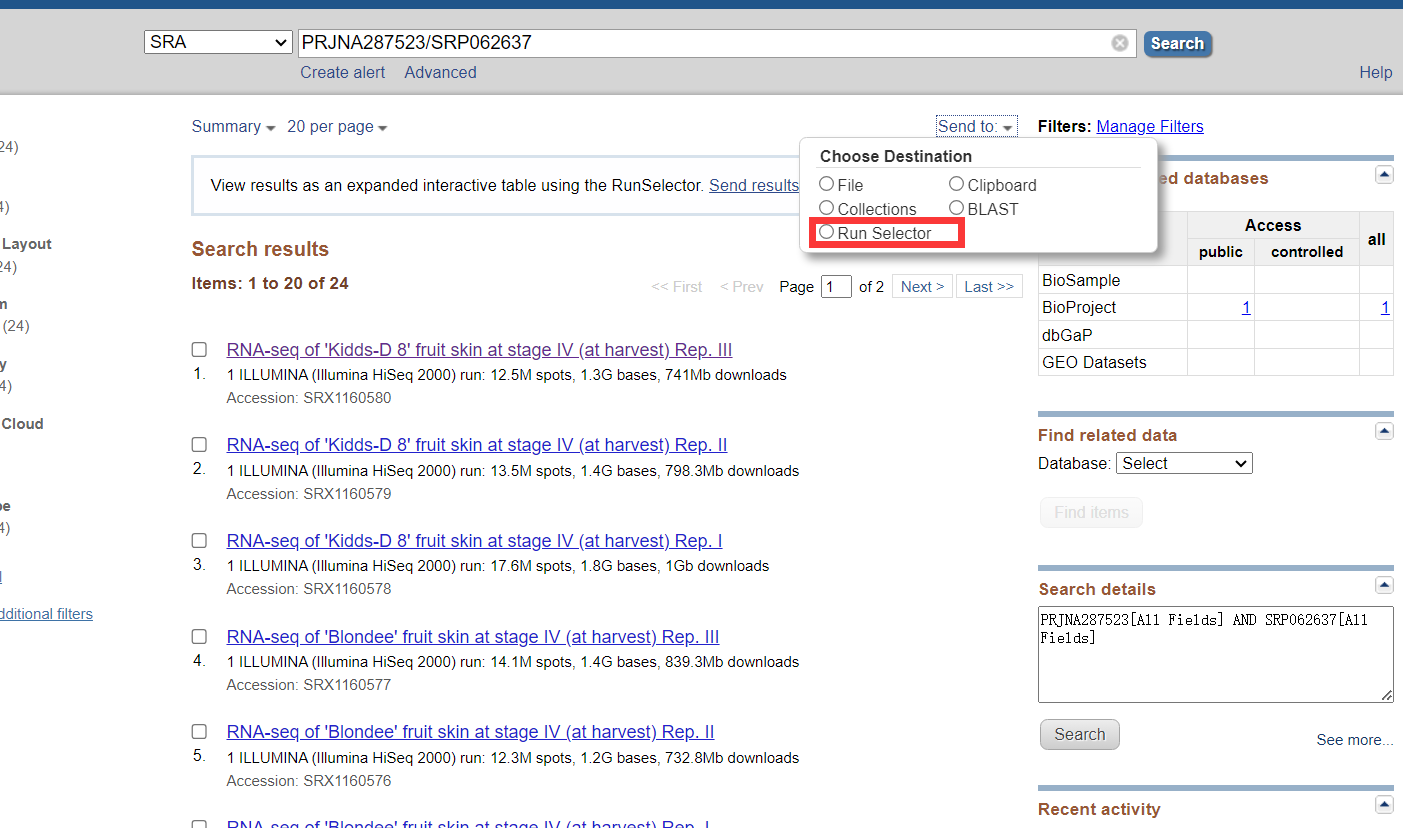
(2)批量下载
获取所有的编号
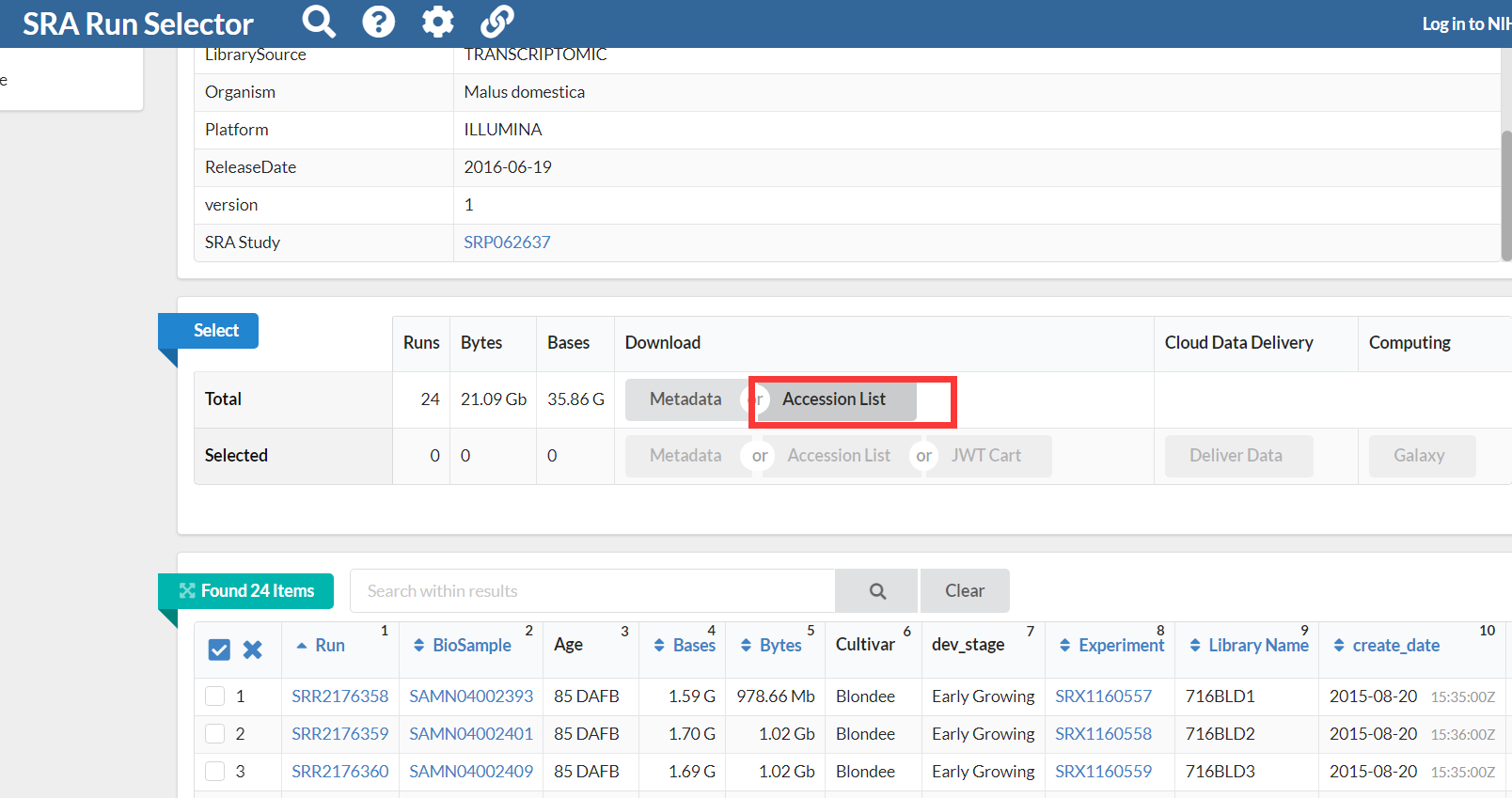
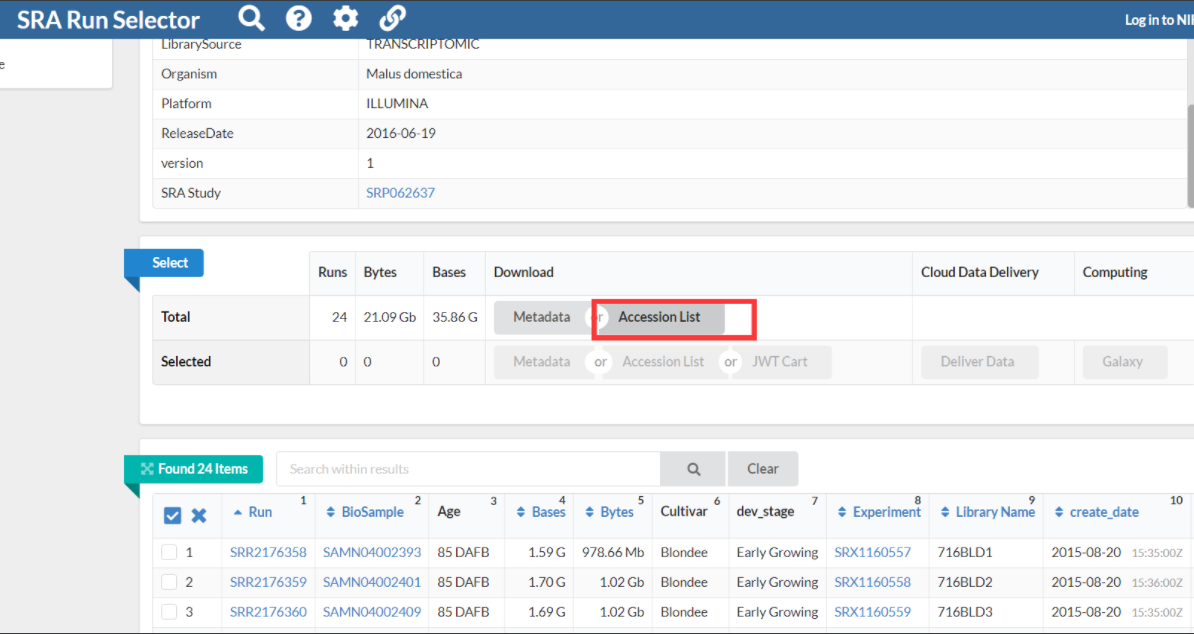
得到SRR_Acc_List.txt,里面包含了24个RNAseq的原始数据文件,增加批量处理的程序,下载这些文件
1 | |
prefetch SRRXXXXX
#下载NCBI里面的项目:SRRXXXXX
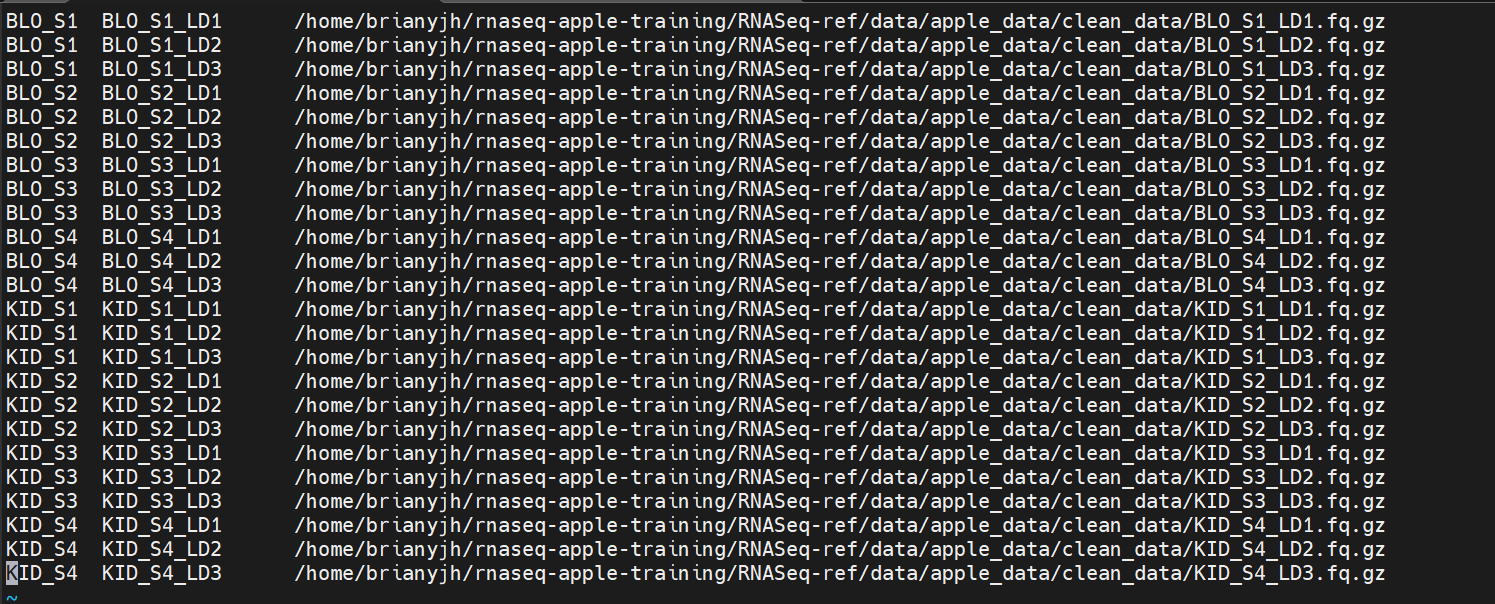
随后批量进行下载 修改一下samples.txt
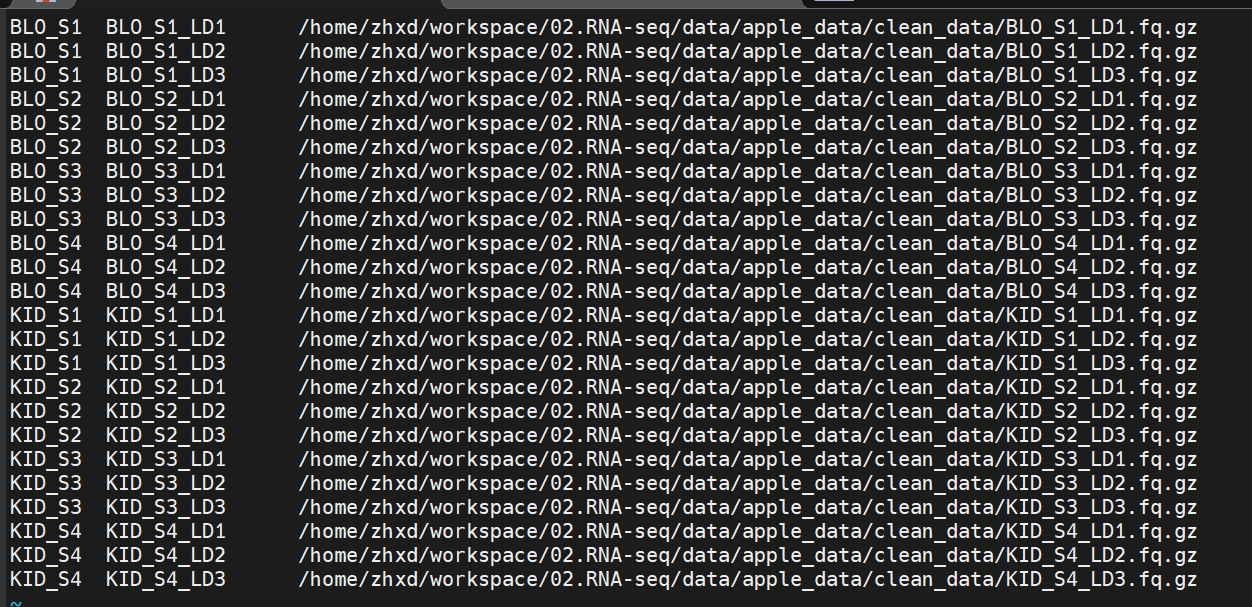
1 | |
:%s/{寻找的片段}/{替换的片段}/g
这里的
/可以替换成#,g表示全部都会被替换
(3)准备参考基因组
参考基因组要准备三个文件:
- 基因组序列(
genome.fasta)- 基因注释(
genes.gtf)- 蛋白序列(
proteins.fasta)
使用苹果参考基因组:https://github.com/moold/Genome-data-of-Hanfu-apple
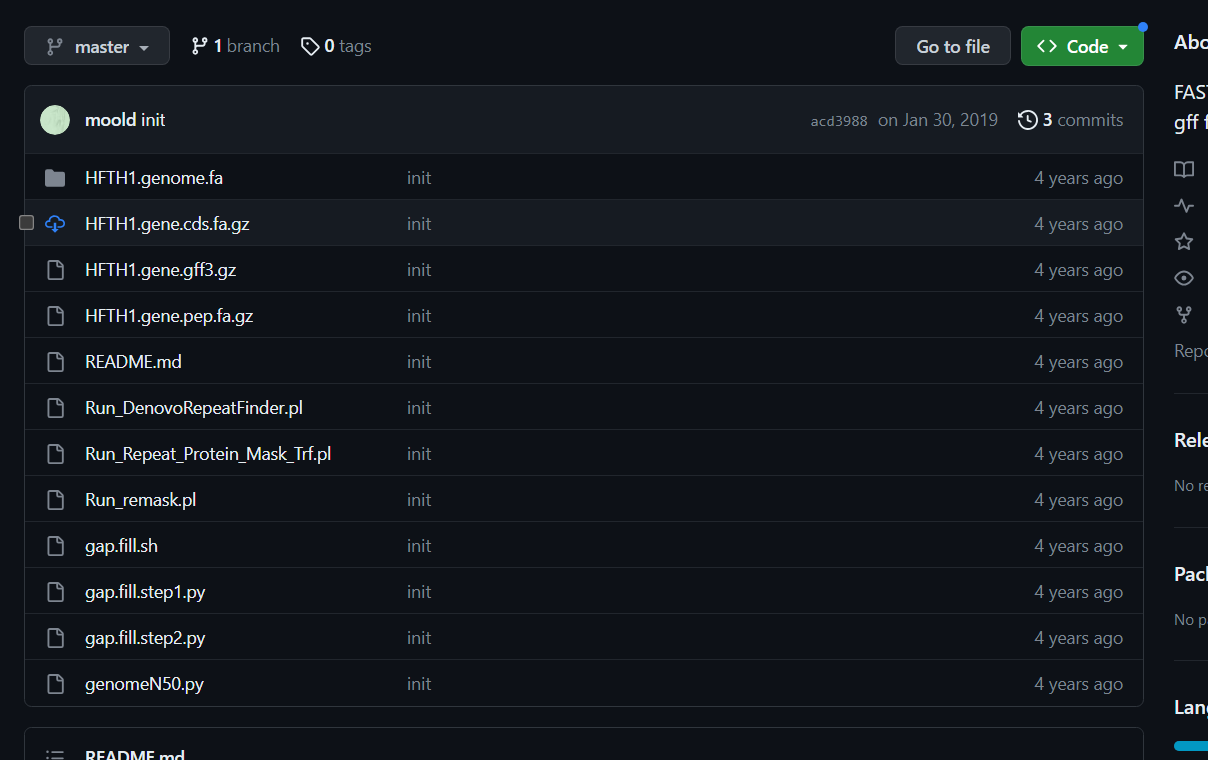
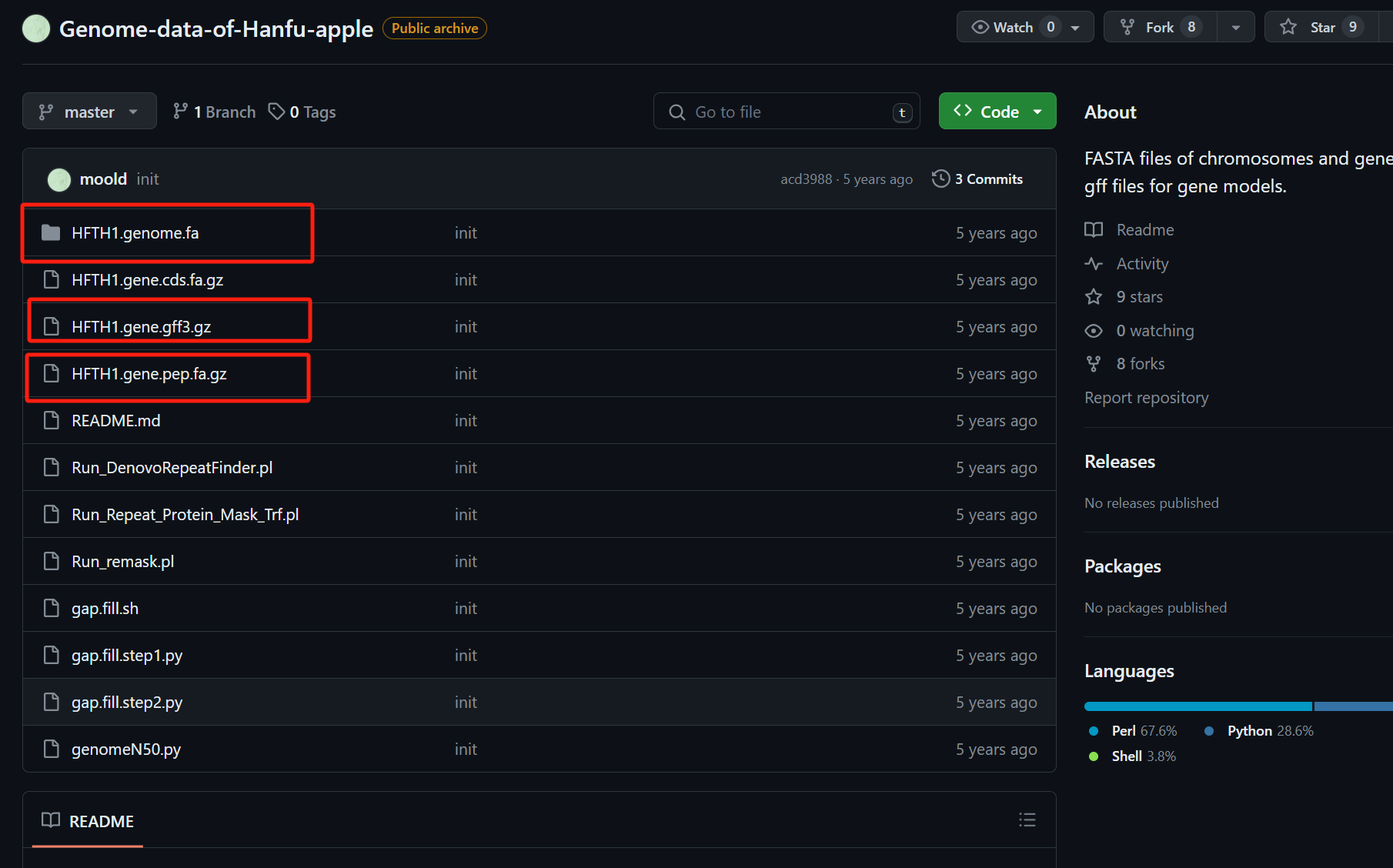
圈中的文件依次是基因组序列文件、基因注释文件(gff文件格式)和蛋白注释文件
基因组序列文件包含了苹果染色体的染色体序列,需要进行合并
1
2cat Chr*.fa.gz > apple_genome.fa.gz
gunzip apple_genome.fa.gz
gff文件通过gffread转化成gtf文件
1
gffread -T -o genes.gtf HFTH1.gene.gff3
(4)比对参考基因组
获取参考基因组后,下一步将RNA-Seq结果比对到参考基因组。
**理论上RNA-Seq的测序数据需要做过滤和质控*
4.1 mapping
构建参考基因组的index
1 | |
这个程序用我的虚拟机跑了得有30分钟,得到index:
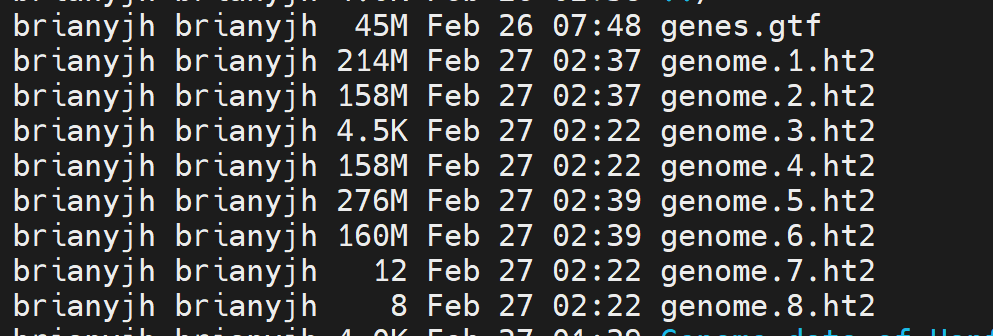
4.2 比对
1 | |
--new-summary 使用新的日志格式 -p {线程数} -x {刚刚创建的index的位置} -U {测序文件,fastq格式} -S 输出文件,sam格式 --rna-strandness { R、dUTP、RF} 普通文库
1 | |
这里进行批处理:
1 | |
如果要并行运行程序,需要在每一个程序后面加一个&,将每一个测序文件进行运行后得到24个比对的文件及相应的日志文件
打开其中一个日志文件BLO_S1_LD1.log,可以看到比对率为76.38%
1
2
3
4
5
6HISAT2 summary stats:
Total reads: 10000000
Aligned 0 time: 2362122 (23.62%)
Aligned 1 time: 7078521 (70.79%)
Aligned >1 times: 559357 (5.59%)
Overall alignment rate: 76.38%
下一步,将sam文件转换成bam文件
1 | |
转成bam文件后,生成对应的index文件 1
2
3samtools index BLO_S1_LD1.bam #生成对应的index文件
awk '{print "samtools index "$2".bam "}' ../data/samples.txt > step4_batch_processing.sh #批处理
nohup sh step4_batch_processing.sh & #后台运行
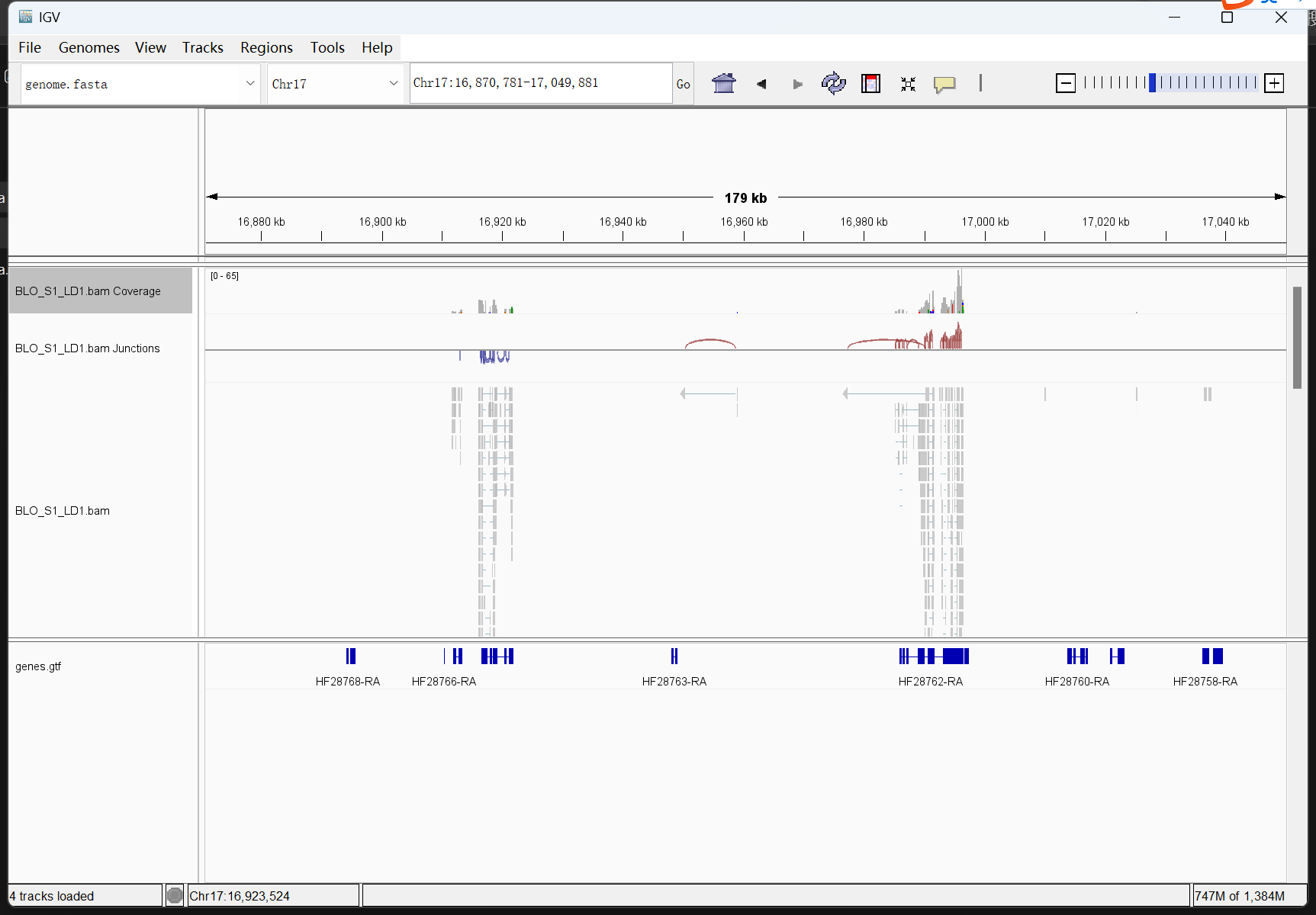
比对完成后,打开IGV软件,通过load genome from file 放入苹果的基因组文件
通过load from file导入gtf文件,同样可以导入bam文件查看比对结果